
Questo sito Web utilizza i cookie in modo che possiamo fornirti la migliore esperienza utente possibile. Le informazioni sui cookie sono memorizzate nel tuo browser ed eseguono funzioni come riconoscerti quando ritorni sul nostro sito Web ed aiutare il nostro team a capire quali sezioni del sito Web ritieni più interessanti e utili.
Le craniostenosi
Cosa sono
Il termine Craniostenosi o Craniosinostosi deriva dal greco e significa chiusura, ristrettezza del cranio.
La Craniostenosi è una delle malformazioni congenite più frequenti, per la quale viene riportata un’incidenza di circa 1 neonato su 2000. La malformazione è caratterizzata dalla prematura saldatura di una o più suture craniche, che comporta una crescita disarmonica del cranio durante i primi due anni di vita.
Il cranio è infatti composto da ossa piatte, separate da suture, che si chiudono normalmente fra il primo ed il terzo anno di vita. Man mano che il cervello dei bambini si sviluppa nei primi due anni di vita, raddoppiando il suo volume, le suture permettono al cranio di espandersi progressivamente e grazie a questa “spinta modellante” si ottiene la crescita armonica della testa.
Quando una o più suture sono chiuse, il cranio si espande lungo le suture che rimangono aperte, con una conseguente progressiva deformazione della sua struttura, che comporta una conformazione disarmonica ed anomala del cranio.
Nei casi più gravi, nei quali sono interessate più suture, oltre alla deformità del cranio si sviluppa anche una aumentata pressione al suo interno (ipertensione endocranica), che può comportare danni al cervello in evoluzione se non viene correttamente e precocemente diagnosticata e trattata. I danni possono colpire la vista, l’udito e le capacità cognitive.
Pertanto è fondamentale che la patologia venga correttamente diagnosticata ed i bambini possano essere sottoposti al corretto trattamento chirurgico entro i primi anni di vita, quando questo permette di ottenere i migliori risultati sia da un punto di vista funzionale sia estetico.
La causa delle Craniostenosi è ancora sconosciuta; per alcune forme più gravi, che implicano l’interessamento di più suture, è stata riconosciuta una alterazione genetica ed è nota una trasmissione familiare, mentre per quanto riguarda le forme che interessano una sola sutura molta ricerca rimane ancora da fare. Sebbene non siano state ancora identificate le alterazioni genetiche è probabile che ve ne siano, in quanto è osservazione frequente una ricomparsa della malformazione nei figli dei soggetti trattatati.
Il trattamento della malformazione è chirurgico ed ha tre finalità:
- alleviare l’ipertensione endocranica, prevenendo i danni cognitivi e vivi che questa può comportare;
- permettere che il cranio accolga e “protegga” la crescita del cervello, coprendolo con una volta ossea;
- cercare di migliorare l’aspetto di questi bambini da un punto di vista estetico. Infatti l’impatto estetico di un volto deformato può comportare un grave danno psicologico ed anche questo è un motivo più che valido per chiedere ai clinici che si applichino con dedizione alla correzione dei difetti del capo e del viso.
Descrizione e classificazione
I geni dei recettori dei fattori di crescita dei fibroblasti (FGFRs) e il gene TWIST sono responsabili delle forme comuni di craniostenosi.
I recettori dei fattori di crescita dei fibroblasti (FGFRs) sono costituiti da 4 diverse proteine, codificate da 4 geni diversi. La biologia dei recettori e dei fattori di crescita risulta molto complessa a causa dell’espressione spazio-temporale specifica, nel corso dello sviluppo embrionale, delle diverse isoforme dei recettori e dei numerosi ligandi.
Dalla più recente letteratura inerente i FGFrs si evidenzia:
- il FGFR4 non pare coinvolto nella patologie inerente la craniostenosi;
- le mutazioni nei FGFRs sono altamente non casuali;
- la quasi totalità delle craniostenosi è associata alla mutazione su FGFR2.

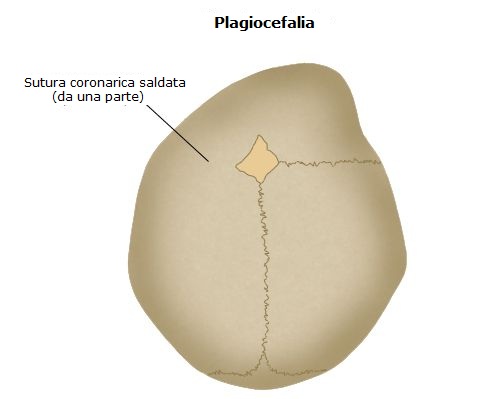
Il coinvolgimento di una sola sutura coronarica avviene nella metà dei casi ed è definita plagiocefalia anteriore (sinistra o destra a seconda del lato in cui si è verificata la saldatura). Ha la prevalenza di circa 1:10.000 bambini nati e la maggioranza dei casi sono sporadici, con una predominanza per il sesso femminile (rapporto M.F = 1:2). È stata descritta un’associazione con l’età paterna avanzata (> 40 anni).
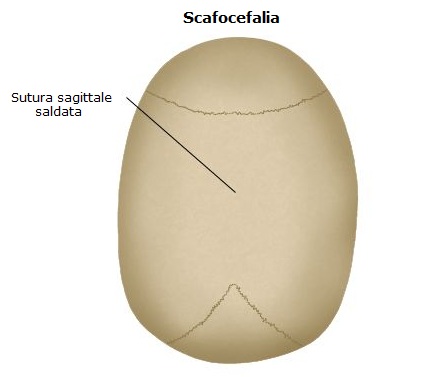
La prematura fusione delle sutura sagittale viene definita dolicocefalia (= testa lunga per il tipico aumento del diametro antero-posteriore) o più comunemente scafocefalia (= la conformazione dello scafo della nave). È la più comune craniostenosi con una prevalenza di circa 1:5.000 bambini nati. Oltre la metà dei casi occorre in forma isolata (cioè non associata ad altre anomalie congenite).
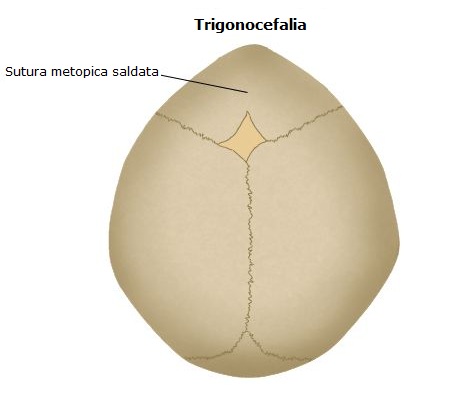
La fusione della sutura metopica avviene nel periodo gestazionale e dà origine alla cosiddetta trigonocefalia; ha una prevalenza alla nascita di circa 1:15.000 con una predominanza nel sesso maschile (M:F=3:1). Circa il 5-6% dei casi sono famigliari. La chiusura anticipata di questa sutura si manifesta con una cresta mediana ed una ristrettezza della fronte, tanto che, e volte, questa sembra appuntita, come un triangolo; gli occhi possono anche essere molto vicini (ipotelorismo).
La plagiocefalia posteriore o sinostosi lambdoidea presenta una prevalenza di 1:15.000 bambini nati con una predominanza nel sesso maschile. È importante distinguere nelle plagiocefalie posteriori, la presenza di un’effettiva sinostosi lambdoidea dalle forme deformazionali, che risultano incrementate dalle recenti prescrizioni pediatriche sulla postura del sonno.

Polisinostosi
Le sinostosi complesse o polisuturali (definite come la contemporanea chiusura di più di una sutura) hanno un’incidenza di circa il 5% dei casi non sindromici (cioè non associata a definiti quadri clinici complessi causati da alterazioni genetiche note).
Il coinvolgimento di due suture avviene in circa i 2/3 dei casi, e l’interessamento di due o più suture in un terzo dei casi. Spesso non interessano suture contigue.
L’interessamento bilaterale della sutura coronale dà origine ad una conformazione cranica corta, definita brachicefalia con riduzione del diametro anteroposteriore del cranio, mentre la chiusura delle due suture lambdoidee comporta una piattezza dell’occipite e viene chiamata pachicefalia. La chiusura di più suture, che comporta una crescita “puntuta” del cranio (verso la fontanella anteriore, che chiude per ultima) viene definita oxicefalia.
In base alla causa invece le craniostenosi possono essere classificate come primarie, cioè non dovute ad altro disordine, oppure secondarie, cioè dovute a patologie metaboliche (ipertiroidismo, rachitismo, mucopolisaccaridosi), ematologiche (talassemia, anemia falciforme), malformazioni (oloprosencefalia, microcefalia, encefalocele), teratogeni (idantoina, retinoidi) o cause iatrogene (sindrome da iperdrenaggio in caso di derivazione liquorale).
Le craniostenosi si presentano spesso come anomalie isolate, oppure possono associarsi ad altre malformazioni o condizioni patologiche all’interno di veri e propri quadri sindromici. Le anomalie associate sono soprattutto a carico degli arti (84%), anomalie dell’orecchio (38%) e difetti cardiaci congeniti (23%), occorrono più frequentemente nelle sinostosi coronali che nelle sinostosi sagittali (Cohen et al 2000). Sono note oltre 90 sindromi con craniostenosi associata di cui circa la metà mostra modalità di trasmissione autosomica dominante o recessiva.
- Autosomica recessiva: due copie del gene sono necessarie per esprimere la malattia, uno ereditato da ciascun genitore, che sono portatori obbligati. Genitori portatori hanno una possibilità su quattro, o 25 per cento, con ogni probabilità la gravidanza, di avere un bambino con craniosinostosi. Maschi e femmine sono ugualmente colpiti.
- Autosomica dominante: un gene è necessario per esprimere la malattia e il gene è passato dai genitori ai figli con un rischio 50/50 per ogni gravidanza. Maschi e femmine sono ugualmente colpiti.
Fattori di rischio
Gli studi epidemiologici sulle craniostenosi hanno evidenziato una predominanza del sesso maschile, in particolare per le sinostosi sagittale e lambdoidea, mentre per le sinostosi coronali si riscontra una predominanza del sesso femminile.
La prematurità (età gestazionale < 37 settimane) e il basso peso alla nascita (< 2500 g) sono riportati quali fattori di rischio. Le craniostenosi appaiono associate con l’incremento dell’età paterna (> 40 anni) in particolare per la sinostosi coronale. Anche l’età materna è stata riportata a tale tipo di craniostenosi, seppur un altro studio non conferma tale dato.
Alcuni studi mostrano un’associazione tra fumo materno in gravidanza e craniostenosi isolate.

Genetica
I geni dei recettori dei fattori di crescita dei fibroblasti (FGFRs) e il gene TWIST sono responsabili delle forme comuni di craniostenosi. I recettori dei fattori di crescita dei fibroblasti (FGFRs) sono costituiti da 4 diverse proteine, codificate da 4 geni diversi. La biologia dei recettori e dei fattori di crescita risulta molto complessa a causa dell’espressione spazio-temporale specifica, nel corso dello sviluppo embrionale, delle diverse isoforme dei recettori e dei numerosi ligandi.
Dalla più recente letteratura inerente i FGFrs si evidenzia:
- il FGFR4 non pare coinvolto nella patologie inerente la craniostenosi;
- le mutazioni nei FGFRs sono altamente non casuali;
- la quasi totalità delle craniostenosi è associata alla mutazione su FGFR2.
Forme sindromiche più frequenti:
In questa forma si riscontra craniostenosi (sutura coronale e in alcuni casi le altre fino alla pansinostosi), ipoplasia del mascellare, proptosi (globi oculari sporgenti perché le cavità orbitarie sono più piccole), fusione delle vertebre cervicali, anomalie dell’articolazione del gomito, idrocefalo.
È presente craniostenosi con turribrachicefalia, ipoplasia del mascellare, palatoschisi, prognatismo e malocclusione, sindattilia di mani e piedi, fusione delle vertebre cervicali, malformazioni encefaliche (agenesia del corpo calloso e del setto pellucido), idrocefalo ed erniazione delle tonsille cerebellari (anomalia di Chiari).
Craniostenosi (brachicefalia o plagiocefalia anteriore), scoliosi della faccia, ptosi palpebrale, strabismo, ipertelorismo, deviazione del setto nasale, sindattilia moderata (solo il 2° e 3° dito di mani e piedi), fusione dei corpi vertebrali.
Si riscontra craniostenosi con turri-brachicefalia, proptosi anche molto importante (l’occhio può anche fuoriuscire dalla cavità orbitaria), ipoplasia del mascellare, ipertelorismo, sindattilia di tessuto molle, fusione delle vertebre cervicali, anchilosi del gomito, idrocefalo, erniazione tonsillare, con o senza minimo ritardo mentale.
Sintomi
Nei neonati con questa malattia, il cambiamento nella forma della testa e del viso può essere evidente.
Sintomatologicamente le craniostenosi vanno classificate in compensate e scompensate.
Le forme compensate presentano un accrescimento del cervello normale senza apparenti segni di compressione del sistema nervoso. I segni clinici più evidenti riguardano principalmente lo scheletro, deformazione cranica, chiusura precoce della fontanella bregmatica, ridotto perimetro cranico e alterazioni facciali.
Le forme scompensate, sono palesemente più gravi, e determinano una sintomatologia d’ipertensione endocranica, che può instaurarsi in modo subdolo e presentare una sintomatologia invalidante. L’ipertensione endocranica comporta una sofferenza del sistema nervoso che si esprime attraverso una serie di sintomi neurologici.
Diagnosi
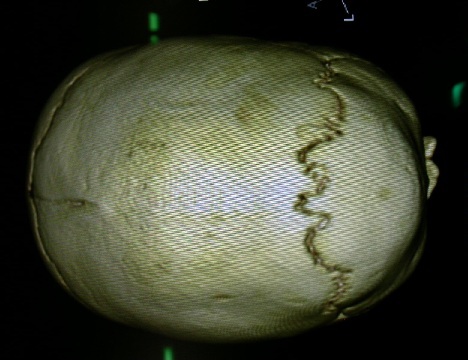
La diagnosi per le craniostenosi viene effettuata sulla base dell’esame obiettivo e viene confermata grazie all’ausilio della TC encefalo con ricostruzioni 3D.
La TC riveste una notevole importanza soprattutto nei casi dubbi in cui non si riesce a distinguere tra una plagiocefalia posizionale e una vera craniostenosi, oppure quando la craniostenosi non è isolata ma sintomo di altre sindromi più complesse.
Nei bambini di età inferiore ai tre mesi riveste notevole importanza la diagnosi precoce mediante ecografia.
Trattamento
La chirurgia è di solito il trattamento principale.
L’obiettivo del trattamento è quello di ridurre la pressione nella testa e correggere le deformità del viso e delle ossa del cranio.
Il periodo ottimale per effettuare la chirurgia è prima dell’anno di età (solitamente tra il 5-9 mese) in quanto le ossa sono ancora molto morbide e facili da lavorare.
La chirurgia può essere necessaria in età molto più precoce a seconda della gravità della malattia.
Sostieni la nostra Onlus
AICRA è una Organizzazione No Profit, quindi ogni donazione tracciabile (effettuata attraverso: bonifici bancari, assegni circolari o bancari intestati ad AiCRA Associazione Craniostenosi recanti la clausola “non trasferibile”, carte di credito, carte di credito prepagate) è deducibile o detraibile secondo le normative vigenti (DL 35/2005; DPR 917/86).
Contattaci per qualsiasi informazione!
